Ipomelanosi di Ito.
##submission.downloads##
Come citare
Abstract
L’ipomelanosi di Ito, è una rara malattia, descritta per la prima volta nel 1952 da Minor Ito con il nome di incontinentia pigmenti achromians, in quanto presenta somiglianze con l’incontinenza pigmentaria di Bloch-Sulzberger. Le è stato dato il nome attuale nel 1973 da Jelinek e coll. La malattia è sporadica, anche se sono noti alcuni rari e discutibili casi familiari, e in almeno il 50% dei casi è legata a mosaicismo cromosomico.
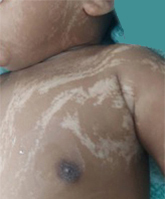
Il fenotipo caratteristico consiste in lesioni ipopigmentate, con differenti dimensioni, distribuzione e forme (linee o vortici), che possono verificarsi unilateralmente o bilateralmente. Tali lesioni colpiscono principalmente il tronco e gli arti, risparmiando generalmente le regioni palmo-plantari e le mucose (4). Le lesioni cutanee possono cambiare nel tempo, progredendo o regredendo. Sono state segnalate numerose anomalie non cutanee associate, tra cui quelle muscolo-scheletriche, neurologiche, oftalmiche, orali, dentali, cardiache, genitali e urinarie. Le anomalie muscolo-scheletriche e del sistema nervoso centrale sono le più frequenti e, sebbene convulsioni, ritardo dello sviluppo neuro-psicomotorio, disturbi dell’andatura e autismo possano coesistere, nessuno di questi disturbi è specifico dell’ipomelanosi di Ito.
