Spettro fenotipico clinico della sindrome di Schimmelpenning-Feuerstein-Mims: una revisione sistematica.
##submission.downloads##
Come citare
Abstract
Background. La sindrome di Schimmelpenning-Feuerstein-Mims (SFMS) rappresenta una rara malattia neurocutanea a mosaico causata da mutazioni post-zigotiche della via di segnalazione RAS-MAPK. È caratterizzata da nevo epidermico-sebaceo lineare associato a un coinvolgimento variabile neurologico, oculare, scheletrico e metabolico. Nonostante siano trascorsi quasi sette decenni dalla descrizione iniziale, i dati fenotipici complessivi restano maldefiniti.
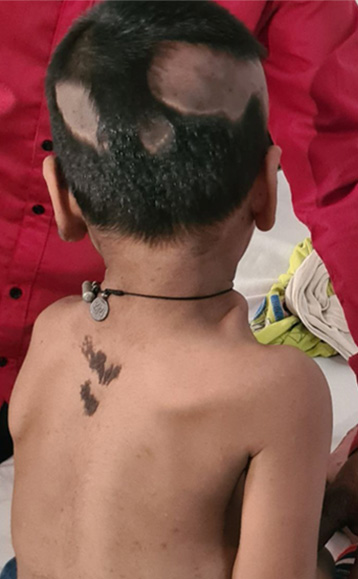
Metodi. Revisione sistematica della letteratura (1957-2025) secondo le linee guida PRISMA. Su 182 record identificati, 28 studi hanno soddisfatto i criteri di inclusione. Un caso indice (maschio di 5 anni con nevo epidermico-sebaceo esteso, displasia scheletrica, grave ipofosfatemia – 0,58 mmol/L –) si aggiunge alla precedente casistica.
Risultati. Il nevo epidermico-sebaceo era presente nel 100% dei casi. Il coinvolgimento extracutaneo comprendeva anomalie neurologiche (50-75%), difetti oculari (40-60%) e anomalie scheletriche (15-30%). Il rachitismo ipofosfatemico mediato da fattore di crescita fibroblastica-23 è un sottogruppo metabolico distinto (5-15%) che richiede una gestione specialistica. Il rischio di carcinoma basocellulare in età pediatrica è <1%, non del 10-20% come si riteneva in passato per un’errata classificazione di trichoblastomi benigni.
Interpretazione. La SFMS presenta un continuum fenotipico che riflette il timing del mosaicismo RAS post-zigotico. Un approccio diagnostico modulare (cute più caratteristiche neurologiche, oculari o scheletriche) dovrebbe sostituire la classica triade ormai obsoleta. Una valutazione multidisciplinare precoce consente una gestione che anticipi le complicanze neurologiche, oftalmologiche e metaboliche.
